
Ibuprofen Synthesis
By McKenna Kilburg '18 & Rachel Tyler '18
CHEM-236: Organic Chemistry II
In my courses (Organic Chemistry I & Organic Chemistry II), most students are exposed to technical writing for the first time. This genre is important because nearly all of the students who complete this course sequence intend to pursue careers that utilize technical writing. To give authenticity to the assignments in my courses, students are instructed to write their “lab reports” as though they were going to submit the paper for publication to the Journal of Organic Chemistry. I found that McKenna and Rachel were able to write this report in a way that was more clear and technically accurate than I have read when reviewing some papers written by professional scientists for journal publications. I think this work deserves to be showcased as an example of the high-level of technical writing Central students are capable of after less than one year of instruction.
-Jay Wackerly
Abstract
The synthesis of ibuprofen was accomplished from isobutylbenzene. The synthetic process included a Friedel-Crafts acylation, reduction, chloride substitution, and Grignard reaction. The products of each step were analyzed using IR and 1H NMR spectroscopy and the final product was additionally confirmed by melting point analysis.
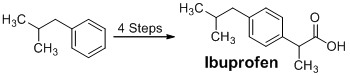
Introduction
Ibuprofen is a nonsteroidal antiinflammatory drug used to relieve pain and reduce swelling, among other common treatments. It was first patented in 1961 by the Boots Pure Chemical Company, and was approved as an over-the-counter drug in 1984.1 Since its creation, ibuprofen has been marketed under several brand names such as Advil® and Motrin®.
In 1992, the BHC Company developed a new, sustainable synthesis that halved the number of synthetic steps (Scheme 1). The first step utilized anhydrous hydrogen fluoride as both a catalyst and solvent, which was then recycled and reused. Additionally, the true brilliance behind the BHC method is the reduced amount of unwanted waste due to the generation of only one molecule of water as the byproduct; this, among other factors, have contributed to a genuinely green synthesis.2
In this report, we describe a five-step synthesis of ibuprofen that mimics the industrial BHC synthesis. Like the BHC method, our synthesis began with a Friedel-Crafts acylation and carbonyl reduction. However, our synthesis utilized a chloride substitution, Grignard formation, and Grignard reaction in the final steps as the industrial method, which uses carbon monoxide (CO) at 500 psi, was avoided due to safety concerns.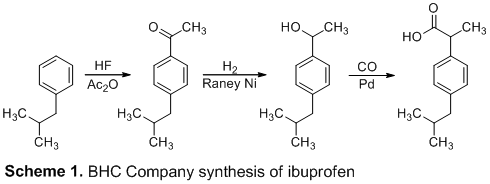
Results & Discussion
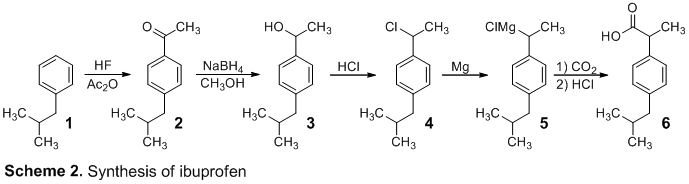 The synthesis of ibuprofen was accomplished through a five-step process shown in Scheme 2. Initially, isobutylbenzene (1) and acetic anhydride, were reacted under Friedel-Crafts acylation conditions to create p-isobutylacetophenone (2). Acetic anhydride and AlCl3 formed a lewis acid complex that produced an acylinium ion, which was then attacked by 1 to form p-isobutylacetophenone (2) through electrophilic aromatic substitution. This product was obtained in 25.6% yield.
The synthesis of ibuprofen was accomplished through a five-step process shown in Scheme 2. Initially, isobutylbenzene (1) and acetic anhydride, were reacted under Friedel-Crafts acylation conditions to create p-isobutylacetophenone (2). Acetic anhydride and AlCl3 formed a lewis acid complex that produced an acylinium ion, which was then attacked by 1 to form p-isobutylacetophenone (2) through electrophilic aromatic substitution. This product was obtained in 25.6% yield.
Ketone 2 was analyzed using IR and 1H NMR spectroscopy. The IR spectrum displayed the presence of Csp2-H, Csp3-H, C=O, and C=C frequencies with peaks at 3026, 2954 cm, 1684, and 1605 cm-1, respectively. This shows the appearance of the appropriate ketone functional group. The 1H NMR spectrum validated the structure of the product. The two doublets integrating to 2 hydrogens at 7.89 and 7.15 ppm suggest 4 aryl hydrogens that are ortho and meta to the acetyl group on the benzene ring. The singlet at 2.52 ppm indicates the α-hydrogens of the acetyl group. The peak at 2.46 ppm suggests 2 methylene hydrogens, and the peak at 1.88 ppm indicates a tertiary hydrogen. The peak at 0.91 ppm suggests 6 methyl hydrogens. However, a high concentration of unreacted isobutylbenzene was observed, and it hindered the experiment in future steps.
In the second step, ketone 2 was reduced with NaBH4 in CH3OH to form 3. The product was obtained in 6.8% yield, most likely due to impure starting material from the first reaction. 3 was confirmed through 1H NMR spectroscopy where a new quartet at 4.81 ppm appeared, indicating the addition of a benzylic hydrogen.
Next, alcohol 3 was substituted by chlorine under acidic conditions to form 4 via an SN1 mechanism. The reaction proceeded smoothly and produced 49.2% yield. The product was analyzed by IR and 1H NMR spectroscopy. The 1H NMR spectrum of 4 does not display the hydroxyl hydrogen at 1.45 ppm, and there was a shift in the benzylic hydrogen peak from 4.81 ppm to 5.09 ppm. The IR spectrum does not contain the broad O-H peak, indicating a replaced alcohol group.
Next, the Grignard reagent, 5, was formed by reacting 4 with magnesium in refluxing ether. Carbon dioxide was bubbled through 5, which permitted the nucleophilic attack of the Grignard reagent onto CO2 to form 6 followed by protonation. The final ibuprofen product, 6, was produced in 24.6% yield. The expected product was confirmed through IR and 1H NMR spectroscopy. The 1H NMR spectrum of 6 displayed a shift in the benzylic hydrogen peak from 5.09 ppm to 3.73 ppm. The IR spectrum suggests the formation of the carboxylic acid with new peaks at 1706 cm-1 indicating the carbonyl bond and a broad peak ranging from 3300 cm-1 to 2400 cm-1 showing the O-H bond. Finally, the observed melting point (68 – 69 °C) is similar to the literature melting point3 (75 – 78 °C) of ibuprofen, indicating the synthesis of impure ibuprofen.
Prior to the loss of product in step 2, the overall yield of the synthesis was 1.74%. This low yield is mostly contributed to the high concentrations of unreacted isobutylbenzene within the first step. The purity could be improved by allowing a longer reaction period or by adding a catalyst. After supplemental stock product was obtained, the remainder of the synthesis yielded 12.1% ibuprofen product.
Conclusion
Ibuprofen was successfully synthesized from the starting materials isobutylbenzene and acetic anhydride through a Friedel-Crafts acylation, carbonyl reduction, chloride substitution, and Grignard reaction. The structure and purity of the product were supported by IR, 1H NMR, and melting point analysis.
Experimental

McKenna Kilburg, silver
General. All starting materials were obtained from commercial suppliers. IR spectra were recorded on a Perkin Elmer FT-IR spectrometer in attenuated total reflection (ATR) mode. The 1H NMR spectra were recorded on a 300 MHz spectrometer. Chemical shifts are expressed in parts per million (δ) using residual TMS as internal standard (δ 0 ppm). Coupling constants, J, are reported in Hertz (Hz), and splitting patterns are designated as s (singlet), d (doublet), t (triplet), q (quartet) and m (multiplet). p-Isobutylacetophenone (2). To a 50 mL round-bottom flask, 5.40 g (60.0 mmol) of AlCl3 and 20 mL of CH2Cl2 were added and cooled on ice. Slowly, 4.03 g (30.0 mmol) of isobutylbenzene and 3.06 g (30.0 mmol) of acetic anhydride were added, and the solution was stirred for 45 min at 0 ºC. The solution was warmed to rt and quenched with 0 ºC 4M HCl solution. The aqueous layer was extracted with dichloromethane (3 × 20 mL). The combined organic layers were washed with 20 mL of 10% NaOH, 20 mL of 50% brine, and 20 mL of H2O, then separated. The solution was dried over Na2SO4, and the solvent was evaporated under a stream of nitrogen gas to produce 1.1 g (6.3 mmol, 26%) of yellow liquid product. 1H NMR (CDCl3, 300 MHz): δ 7.88 (d, 2H, J = 7.5), 7.14 (d, 2H, J = 6.3 Hz), 2.58 (s, 3H,), 2.47 (d, 2H, J = 6.6 Hz), 1.85 (br, m, 1H), 0.903 (d, 6H, J = 6.0 Hz). IR (ATR): 3027 (νCsp2-H, m), 2954 (νCsp2-H, m), 1684 (νC=O, m), 1605 (νC=C, m) cm-1.
1-(4-Isobutylphenyl) ethanol (3). In a separatory funnel, 1.23 mL (6.28 mmol) of p-isobutylacetophenone was dissolved in 6.0 mL of methanol. Quickly, 0.237 g (13.2 mmol) of NaBH4 was added to the solution and allowed to react for 10 min. 20 mL of 10% HCl were added and the aqueous layer was extracted with petroleum ether (3 × 5 mL). The combined organic layers were dried over Na2SO4, and the solvent was evaporated under a stream of nitrogen gas to yield 76 mg (0.43 mol, 6.8%) of yellow liquid product. 1H NMR (CDCl3, 300 MHz): δ 7.25 (d, 2H, J = 7.8 Hz), 7.10 (d, 2H, J = 7.8 Hz), 4.81 (q, 1H, 6.0 Hz), 2.45 (d, 2H, J = 6.6 Hz), 2.27 (s, 1H), 1.84 (m, 1H), 1.45 (d, 3H, J = 6.0 Hz), 1.90 (d, 6H, J = 6.0 Hz).
1-Chloro-1-(4- isobutylphenyl)ethane (4). A 1.0 mL (5.6 mmol) solution of 1-(4-isobutylphenyl)ethane in 20 mL of 12 M HCl was added to a separatory funnel. The funnel was shaken for 3 min, and the product was extracted with petroleum ether (3 × 5 mL). The combined product was dried over Na2SO4, and the solvent was evaporated under nitrogen gas to yield 534 mg (2.71 mmol, 49.2%) clear liquid product. 1H NMR (CDCl3, 300 MHz): δ 7.32 (d, 2H, J = 8.1 Hz), 7.12 (d, 2H, J = 7.8 Hz), 5.09 (q, 1H, J = 6.9 Hz), 2.46 (d, 2H, J = 7.2 Hz), 1.85 (m, 4H), 0.898 (d, 6H, J = 6.6 Hz). IR (ATR): 3020 (νCsp2-H, m), 2954 (νCsp2-H, m), 1511 (νC=C, m) cm-1.
2-(4-Isobutylphenyl) propanoic acid (6). To an oven-dried, 50 mL roundbottom flask, 534 mg (2.71 mmol) of 1-chloro-1-(4- isobutylphenyl)ethane, 20 mL of THF, 1.06 g (43 mmol) of magnesium, and a drop of 1,2-dibromomethane were added. The solution was placed in a reflux condenser and heated under reflux 30 min after foaming was apparent. After cooling to rt, 1 L of CO2 was bubbled through the resulting product, 1-(4-isobutylphenyl) ethylmagnesium chloride, and decanted in a separatory funnel with 10 mL of petroleum ether and 16 mL of 10% HCl. The aqueous phase was extracted with petroleum ether (2 × 10 mL) and the organic layers were combined and extracted into 5% NaOH (2 × 8 mL). The solution was acidified with 20 mL of 10% HCl. Ibuprofen was extracted with petroleum ether (3 × 10 mL), dried over Na2SO4, and the solvent was evaporated under nitrogen gas to yield 138 mg (0.670 mmol, 25%) of solid, white product. 1H NMR (CDCl3, 300 MHz): δ 7.22 (d, 2H, J = 8.1 Hz), 7.11 (d, 2H, J = 8.1 Hz), 3.72 (q, 1H, J = 7.2 Hz), 2.46 (d, 2H, J = 7.2 Hz), 1.84 (m, 1H), 1.51 (d, 3H, J = 7.2 Hz), 0.881 (d, 6H, J = 6.6 Hz). IR (ATR): 3014 (νCsp2-H, m), 2954 (νCsp2-H, m), 1706 (νC=O, m), 1512 (νC=C, m) cm-1; m.p.: 68 – 69 °C (75-77 °C lit.3).
Works Cited
(1) Ibuprofen: A Case Study in Green Chemistry. http://www.ch.ic.ac.uk/ (accessed 4/7/16).
(2) Green Chemistry | EPA. https://www.epa.gov/greenchemistry/ (accessed 4/8/16).
(3) Ibuprofen | The PubChem Project. https://pubchem.ncbi.nlm.nih.gov (accessed 4/8/16).